


Background
Hemophagocytic lymphohistiocytosis (HLH) is an increasingly recognized life-threatening syndrome characterized by excessive inflammation and immune dysregulation. However, little is known about HLH presenting in neonates.
Methods
To describe the phenotype, etiology, and efficacy of therapy for infants presenting within 28 days of birth and diagnosed with HLH prior to 90 days of life, a retrospective multi-center study was conducted identifying 13 previously unreported cases from 8 centers. A systematic literature review identified 58 additional cases that were sufficiently similar to allow pooling for meta-analysis (n=71).
Results
Presenting symptoms and laboratory findings of HLH were non-specific and indistinguishable from other neonatal critical illnesses. More than 85% of patients demonstrated hepatomegaly, splenomegaly, hyperferritinemia, transaminitis, hypoalbuminemia, and severe thrombocytopenia. Notably, liver dysfunction appears more common in neonatal HLH compared to published reviews of HLH cases across the age spectrum.
Genetic testing was pursued in 60% of the cohort with pathogenic variants identified in a third of patients (PRF1 in 15, UNC13D in 8). Ten additional cases occurred with a family history of HLH, for a total of 46% (33/71) of cases of presumed familial HLH. Seventeen cases (24%) represented secondary HLH with an identified trigger for immune dysregulation (e.g. infection or non-HLH disease) in the absence of an HLH-associated genetic mutation or family history. Twenty-one (30%) neonatal HLH cases were of unknown etiology.
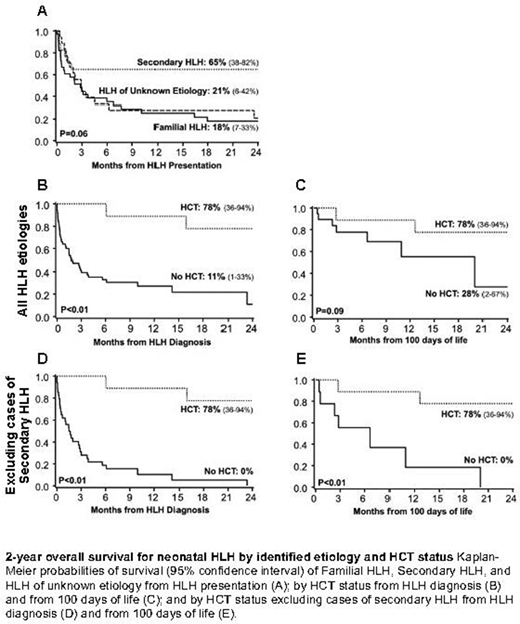
Two-year survival from presentation was low at 27%, with a trend toward superior 2-year overall survival for those with secondary HLH at 65% (unknown etiology 21%, familial HLH 18%, p=0.06; Figure). Ten patients (n=6 familial HLH, n=4 unknown etiology) underwent allogeneic hematopoietic cell transplantation (HCT) at a median of 4.8 months of age. HCT was associated with improved 2-year overall survival (OS) from HLH diagnosis (78% with HCT versus 11% without HCT, p<0.01). Infants proceeding to HCT trended toward older age at diagnosis with less severe lung and liver disease. Suspecting disease severity and/or lethality to confound treatment choice and prohibit proceeding to HCT, a landmark analysis of survival was conducted including only those surviving to 100 days of life. While this landmark analysis attenuated the survival benefit of HCT (78% with HCT versus 28% without HCT, p=0.09), the attenuation was driven by inclusion of secondary HLH cases (2-year OS 78% with HCT versus 0% without HCT when secondary HLH excluded, p<0.01).
Conclusion
Liver dysfunction in a severely ill neonate should prompt consideration of HLH as an etiology. Our study supports consideration of HCT for any neonate with HLH, except those with an identified trigger and absence of an HLH-associated gene mutation prompting classification as secondary HLH. Early lethality combined with superior HCT outcomes when HLH is well-controlled additionally highlight the need for novel HLH therapies to bridge to HCT. Further studies to better elucidate details of pre-HCT therapy, HCT conditioning regimens and how best to navigate multi-organ failure at HLH presentation are needed.
No relevant conflicts of interest to declare.
This icon denotes a clinically relevant abstract

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal